Les prestations pouvant être réalisées via notre unité couvrent : l’extraction, le génotypage, les analyses de transcriptome, le séquençage NGS et les analyses bioinformatiques et statistiques de vos données. Nous sommes CSPro pour le séquençage via les équipements Illumina et pour les captures SureSelect Agilent.
Extractions
Méduse d’ADN en suspension /CNRS Photothèque
Génotypage, Expression et kinome

Différents formats de puces Illumina pour le génotypage ou les études d’expression.
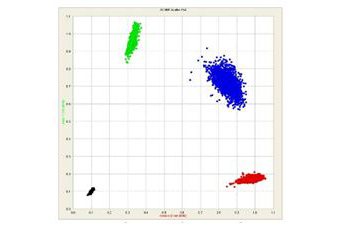
SNP genotyping scatter plot : répartition des génotypes sous forme de clusters après analyse
Séquençage NGS
Préparation de librairies CoDE-seq, WES, WGS, RNA seq, ChIP seq :
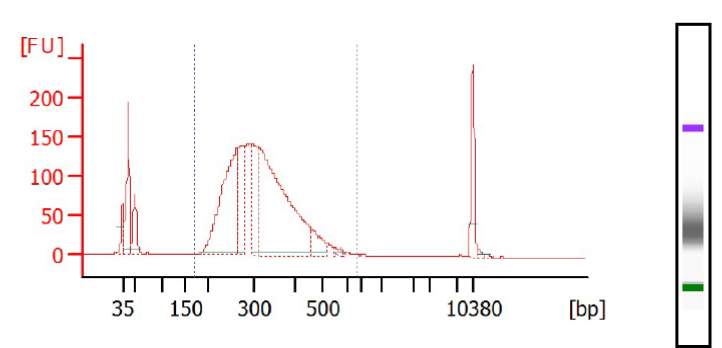
Profils de librairies de Whole Exome (KAPA HyperCap With HyperExome Probes) obtenus via Bioanalyser, puces DNA High Sens – Ces librairies ont été fragmentées et préparées via Sciclone® G3 NGSx iQ™ Workstation (PerkinElmer®). Elles sont prêtes pour le séquençage ciblé via nos séquenceurs Illumina (NovaSeq ou NextSeq).
Nos séquenceurs de nouvelle génération (NGS) : NovaSeq6000Dx, NextSeq500, MiSeq – Illumina
Nous avons acquis le NovaSeq 6000Dx en décembre 2022 et le NovaSeq X+ en mai 2023, premiers équipements de ces types installés en France, tout comme précédemment en juin 2017 notre NovaSeq 6000! Nous possédons également un NextSeq500 et un MiSeq.
L’ensemble de nos équipements NGS nous permettent de vous proposer plusieurs débits de séquençage : le très haut débit, le haut débit et le moyen débit, et ainsi nous adapter à vos projets. Quel que soit le mode choisi, tout type de librairie est susceptible d’être séquencé sur les HiSeq : Exomes, small RNA, total RNA, mRNA, MethylSeq, ChipSeq, amplicons, genomes, RAD…
Nous sommes certifié pour le séquençage via les équipements Illumina.

L’outil Sequencing analysis viewer (Illumina) permet de visualiser les paramètres qualité de séquençage de nos NovaSeq 6000Dx, NextSeq ou MiSeq en cours de run ou à la fin d’un run.
A 1 : schéma d’une flow cell (ici 4 lanes d’un run NovaSeq S4) avec code de couleur reflétant la qualité du séquençage d’une flow cell ; 2 : données par cycle de séquençage, chaque base est représentée par une couleur ; 3 : QScore distribution, permet de visualiser le nombre de reads par score de qualité, seuls les reads passant les filtres qualité sont comptabilisés ; 4 : données par lane, (en nombre de clusters / mm²), 5 : QScore Heatmap : permet de visualiser le QScore par cycles. B visualisation en détail d’une section de flow cell. Différents paramètres de qualité peuvent ainsi être analysés (densité de clusters ayant passé les filtres, taux d’erreur (déterminé par l’alignement avec le PhiX,…)
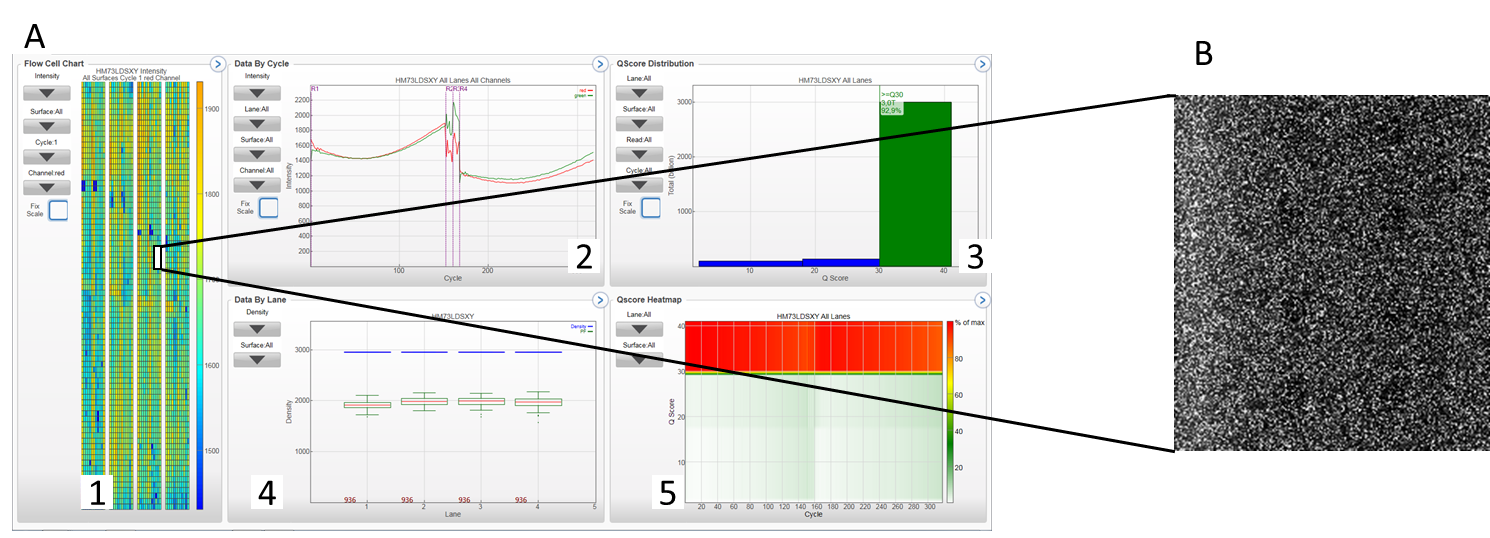
Vue d’ensemble d’un run de séquençage / Image d’une section de flow cell
Analyses bioinformatiques et statistiques

Alignements de séquences issues de séquenceur haut débit, après utilisation d’outil de type Integrative Genomics Viewer